

Romano-Ward症候群
LQT1 KCNQ1 11p15.5
LQT2 KCNH2 7q35-36
LQT3 10.5 3p21-23
LQT4 Ankyrin-B 4q25-27
LQT5 KCNE1 21q22.1-q22.2
LQT6 KCNE2 21q22.1-q22.2
LQT7 KCNJ2 17q23.1
LQT8 CACNA1C 12p13.3
LQT9 CAV3 3p25
LQT10 SCN4B 11q23.3
LQT11 AKAP-9 7q21-q22
LQT12 SNTA1 20q11.2
Jervell & Lange-Nielsen症候群
JLN1 KCNQ1(homozygous) 11p15.5
JLN2 KCNE1(homozygous) 21q22.1-q22.2
Brugada症候群
BrS1 SCN5A 3p21-24
BrS2 CACNA1C 12p13.3
BrS3 CACNB2 10p12.33
BrS4 GPD1-L 3p21
BrS5 SCN1B 19q13.1
BrS6 KCNE3 11q13-q14
カテコラミン誘発性多形性心室頻拍
RyR2 RyR2 1q42-q43
LQT1 KCNQ1 11p15.5
LQT2 KCNH2 7q35-36
LQT3 10.5 3p21-23
LQT4 Ankyrin-B 4q25-27
LQT5 KCNE1 21q22.1-q22.2
LQT6 KCNE2 21q22.1-q22.2
LQT7 KCNJ2 17q23.1
LQT8 CACNA1C 12p13.3
LQT9 CAV3 3p25
LQT10 SCN4B 11q23.3
LQT11 AKAP-9 7q21-q22
LQT12 SNTA1 20q11.2
Jervell & Lange-Nielsen症候群
JLN1 KCNQ1(homozygous) 11p15.5
JLN2 KCNE1(homozygous) 21q22.1-q22.2
Brugada症候群
BrS1 SCN5A 3p21-24
BrS2 CACNA1C 12p13.3
BrS3 CACNB2 10p12.33
BrS4 GPD1-L 3p21
BrS5 SCN1B 19q13.1
BrS6 KCNE3 11q13-q14
カテコラミン誘発性多形性心室頻拍
RyR2 RyR2 1q42-q43
蛋白遺伝子遺伝様式主な表現型
心筋βミオシン重鎖MYH7 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症,左室緻密化障害
心筋トロポニン-T TNNT2 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症
α- トロポミオシンTPM1 AD 肥大型心筋症,拡張型心筋症
心筋ミオシン結合蛋白-C MYBPC3 AD 肥大型心筋症,拡張型心筋症
心室型ミオシン必須軽鎖MYL3 AD 肥大型心筋症
心室型ミオシン調節軽鎖MYL2 AD 肥大型心筋症
心筋トロポニン-I TNNI3 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症
心筋α- アクチンACTC AD 肥大型心筋症,拡張型心筋症,左室緻密化障害
タイティンTTN AD 肥大型心筋症,拡張型心筋症
心筋トロポニン-C TNNC1 AD 肥大型心筋症,拡張型心筋症
心筋α- ミオシン重鎖MYH6 AD 肥大型心筋症
筋肉LIM- ドメイン蛋白CSRP3 AD 肥大型心筋症,拡張型心筋症
カベオリン-3 CAV3 AD 肥大型心筋症
メタビンキュリンVCL AD 肥大型心筋症,拡張型心筋症
ジャンクトフィリン-2 JPH-2 AD 肥大型心筋症
オブスキュリンOBSCN AD 肥大型心筋症
デスミンDES AD 拡張型心筋症,拘束型心筋症,ミオパチー
α- アクチニンACTN2 AD 拡張型心筋症
サイファー,ZASP LDB3 AD 拡張型心筋症,左室緻密化障害
ホスフォランバンPLB AD 拡張型心筋症,肥大型心筋症
K-ATPチャネルABCC9 AD 拡張型心筋症
心筋NaチャネルSCN5A AD 拡張型心筋症
αBクリスタリンCRYAB AD 拡張型心筋症,肥大型心筋症
FHL2 FHL2 AD 拡張型心筋症
ラミニンα4 LMNA4 AD 拡張型心筋症
マイオファラディンMYPN XR 拡張型心筋症
ジストロフィンDMD XR 拡張型心筋症,筋ジストロフィー(Duchenne,Becker)
ラミン A/C LMNA AD 拡張型心筋症,左室緻密化障害,筋ジストロフィー(Emery-Dreifuss)
δ - サルコグリカンSAGD AD 拡張型心筋症,筋ジストロフィー(limb girdle)
エメリンEMD XR 拡張型心筋症
タファディン,G4.5 TAZ XR 左室緻密化障害,拡張型心筋症,Borth病
フクチンFKTN XR 拡張型心筋症
デスモプラキンDSP AR 不整脈源性右室心筋症,拡張型心筋症
プラコグロビンJUP AR,AD 不整脈源性右室心筋症,拡張型心筋症
プラコフィリンPKP2 AD 不整脈源性右室心筋症
TGFβ3 TGFB3 AD 不整脈源性右室心筋症
リアノジン受容体-2 RYR2 AD 不整脈源性右室心筋症
デスモグレイン3 DSG3 AD 不整脈源性右室心筋症
α- ディストロブレビンDTNA AD 左室緻密化障害
AMP活性化プロテインキナーゼPRKAG2 AD 肥大型心筋症,WPW症候群
α- ガラクトシダーゼGALA XR Fabry病
α1,4- グルコシダーゼGAA AR Pompe病
ライソゾーム膜蛋白2 LAMP2 XR Danon病
心筋βミオシン重鎖MYH7 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症,左室緻密化障害
心筋トロポニン-T TNNT2 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症
α- トロポミオシンTPM1 AD 肥大型心筋症,拡張型心筋症
心筋ミオシン結合蛋白-C MYBPC3 AD 肥大型心筋症,拡張型心筋症
心室型ミオシン必須軽鎖MYL3 AD 肥大型心筋症
心室型ミオシン調節軽鎖MYL2 AD 肥大型心筋症
心筋トロポニン-I TNNI3 AD 肥大型心筋症,拡張型心筋症,拘束型心筋症
心筋α- アクチンACTC AD 肥大型心筋症,拡張型心筋症,左室緻密化障害
タイティンTTN AD 肥大型心筋症,拡張型心筋症
心筋トロポニン-C TNNC1 AD 肥大型心筋症,拡張型心筋症
心筋α- ミオシン重鎖MYH6 AD 肥大型心筋症
筋肉LIM- ドメイン蛋白CSRP3 AD 肥大型心筋症,拡張型心筋症
カベオリン-3 CAV3 AD 肥大型心筋症
メタビンキュリンVCL AD 肥大型心筋症,拡張型心筋症
ジャンクトフィリン-2 JPH-2 AD 肥大型心筋症
オブスキュリンOBSCN AD 肥大型心筋症
デスミンDES AD 拡張型心筋症,拘束型心筋症,ミオパチー
α- アクチニンACTN2 AD 拡張型心筋症
サイファー,ZASP LDB3 AD 拡張型心筋症,左室緻密化障害
ホスフォランバンPLB AD 拡張型心筋症,肥大型心筋症
K-ATPチャネルABCC9 AD 拡張型心筋症
心筋NaチャネルSCN5A AD 拡張型心筋症
αBクリスタリンCRYAB AD 拡張型心筋症,肥大型心筋症
FHL2 FHL2 AD 拡張型心筋症
ラミニンα4 LMNA4 AD 拡張型心筋症
マイオファラディンMYPN XR 拡張型心筋症
ジストロフィンDMD XR 拡張型心筋症,筋ジストロフィー(Duchenne,Becker)
ラミン A/C LMNA AD 拡張型心筋症,左室緻密化障害,筋ジストロフィー(Emery-Dreifuss)
δ - サルコグリカンSAGD AD 拡張型心筋症,筋ジストロフィー(limb girdle)
エメリンEMD XR 拡張型心筋症
タファディン,G4.5 TAZ XR 左室緻密化障害,拡張型心筋症,Borth病
フクチンFKTN XR 拡張型心筋症
デスモプラキンDSP AR 不整脈源性右室心筋症,拡張型心筋症
プラコグロビンJUP AR,AD 不整脈源性右室心筋症,拡張型心筋症
プラコフィリンPKP2 AD 不整脈源性右室心筋症
TGFβ3 TGFB3 AD 不整脈源性右室心筋症
リアノジン受容体-2 RYR2 AD 不整脈源性右室心筋症
デスモグレイン3 DSG3 AD 不整脈源性右室心筋症
α- ディストロブレビンDTNA AD 左室緻密化障害
AMP活性化プロテインキナーゼPRKAG2 AD 肥大型心筋症,WPW症候群
α- ガラクトシダーゼGALA XR Fabry病
α1,4- グルコシダーゼGAA AR Pompe病
ライソゾーム膜蛋白2 LAMP2 XR Danon病
疾患名心血管系異常心血管系以外の表現型疾患遺伝子,変異蛋白遺伝子座
Kabuki 症候群大動脈縮窄,大動脈二尖弁,僧帽弁逸脱,心室中隔欠損,肺動脈狭窄,大動脈狭窄,僧帽弁狭窄,Fallot四徴症, 単心室, 両大血管右室起始,大血管転位顔貌異常,精神運動発達遅滞,皮膚紋理異常,骨格異常(脊柱側彎,股関節形成障害,小指短縮),難聴
Unknown Sporadic
LEOPARD症候群肺動脈狭窄,房室ブロック,肥大型心筋症多発性黒子,眼間開離,外陰部
異常,精神遅滞,成長障害,難聴
PTPN11,KRAS
SOS1,RAF1
12q24.1,
12p12.1
2p22-p21,3p25
Marfan症候群大動脈拡大,房室弁閉鎖不全,僧帽弁逸脱,大動脈弁輪拡大,解離性大動脈瘤,肺動脈拡張,肺動脈弁閉鎖不全高身長,水晶体脱臼,近視,青色強膜,脊柱彎曲,漏斗胸,クモ状指,関節の過伸展,長い手足,
FBN1(Fibrillin)
TGFBR1,2
15q21.1
9q33-q34,
3p24.1
Leigh脳症,NARP症候群
肥大型心筋症進行性精神運動発達障害,痙攣,小脳失調,哺乳嚥下障害,筋緊張低下,視神経萎縮
mitochrondrial loci mitochondrial
DNA
MERRF症候群心筋症ミオクローヌス,癲癇,小脳失調,筋緊張低下,知的退行,低身長
MTTK mitochondrial
DNA
筋緊張性ジストロフィー
刺激伝導系障害,心筋症,僧帽弁閉鎖不全筋強直,筋変性,白内障,眼瞼下垂
DMPK,ZNF9 19q13.2 3q13.3
Noonan症候群肺動脈狭窄,肥大型心筋症,心房中隔欠損翼状頚,低身長,発達遅滞,鳩胸,
漏斗胸,眼瞼下垂,出血傾向,血小板機能異常
PTPN11,KRAS,
SOS1,RAF1
12q24.1,
12p12.1,
2p22-p21,3p25
骨形成不全僧帽弁逸脱,大動脈弁閉鎖不全,大動脈拡大
骨脆弱性,多発骨折,難聴,青色強膜,下肢の彎曲変形および短縮,成長障害,顔貌異常
COL1A1
COL1A2
17q21.33
7q21.3
トリソミー13 心室中隔欠損,動脈管開存,心房中隔欠損
精神発達遅滞,全前脳胞症,小頭症,前額部傾斜,難聴,耳介変形,揺り椅子状足底,多指趾
Multiple Trisomy 13
Pompe病グリコーゲン蓄積による心筋肥大
先天性代謝異常,筋力低下,肝腫大,巨舌
GAA(Lysosomal Alpha-Glucosidase)
17q25
Rubinstein-Taybi 症候群
多岐の先天性心疾患,左心低形成発達障害,顔貌異常,多毛,眼瞼裂斜下,眼間開離,上顎低形
成,前額部突出,低身長,幅広い母指趾
CREBBP(CREB-binding protein)
16p13.3
Treacher-Collins 症候群
心室中隔欠損,動脈管開存,心房中隔欠損耳介形成異常,難聴,下顎形成不全,頬骨形成不全,脈絡膜欠損,両側下眼瞼部欠損,口蓋裂,後鼻腔閉鎖
TCOF1(Treacle protein)
5q32
結節性硬化症心臓腫瘍(横紋筋腫),不整脈腫瘍,痙攣,顔面血管線維種,
白斑,カフェオレ斑,骨硬化,腎形成異常,精神発達遅滞,自閉症
TSC1(Hamartin),TSC2(Tuberin)9q34
16p13.3
Turner症候群大動脈縮窄,大動脈二尖弁,左心低形成,心房中隔欠損,心室中隔欠損
低身長,翼状頚,盾状胸,毛髪線の低位,卵巣形成異常,腎形成異常,難聴
Multiple Unisomy X(45,X)
VACTERL連合心室中隔欠損,心房中隔欠損,動脈管開存脊柱の異常,鎖肛,気管食道瘻,
橈骨異形成,四肢の異常,腎泌尿器の異常
Numerous loci Unknown
22q11.2欠失症候群
大動脈離断,総動脈幹遺残,肺動脈閉鎖を伴うFallot四徴症,右側大動脈弓,鎖骨下動脈起始
異常,心室中隔欠損円錐動脈幹異常顔貌,鼻咽腔不全口蓋裂,胸腺低形成,副甲状腺機能低下,低カルシウム血症,易感染性,鎖肛,精神発達遅滞,精神障害,血小板減少症TBX1,UFD1L,
del 22q11.2
Williams 症候群大動脈弁上狭窄,肺動脈弁上狭窄,末梢性肺動脈狭窄,大動脈狭窄,肺動脈狭窄,心筋症
精神発達遅延,妖精様顔貌,虹彩の放射線模様,高カルシウム血症,不正咬合,視空間認知障害,関節拘縮,筋緊張亢進,学習障害,視覚認知障害ELN(Elastin) 7q11.23
Kabuki 症候群大動脈縮窄,大動脈二尖弁,僧帽弁逸脱,心室中隔欠損,肺動脈狭窄,大動脈狭窄,僧帽弁狭窄,Fallot四徴症, 単心室, 両大血管右室起始,大血管転位顔貌異常,精神運動発達遅滞,皮膚紋理異常,骨格異常(脊柱側彎,股関節形成障害,小指短縮),難聴
Unknown Sporadic
LEOPARD症候群肺動脈狭窄,房室ブロック,肥大型心筋症多発性黒子,眼間開離,外陰部
異常,精神遅滞,成長障害,難聴
PTPN11,KRAS
SOS1,RAF1
12q24.1,
12p12.1
2p22-p21,3p25
Marfan症候群大動脈拡大,房室弁閉鎖不全,僧帽弁逸脱,大動脈弁輪拡大,解離性大動脈瘤,肺動脈拡張,肺動脈弁閉鎖不全高身長,水晶体脱臼,近視,青色強膜,脊柱彎曲,漏斗胸,クモ状指,関節の過伸展,長い手足,
FBN1(Fibrillin)
TGFBR1,2
15q21.1
9q33-q34,
3p24.1
Leigh脳症,NARP症候群
肥大型心筋症進行性精神運動発達障害,痙攣,小脳失調,哺乳嚥下障害,筋緊張低下,視神経萎縮
mitochrondrial loci mitochondrial
DNA
MERRF症候群心筋症ミオクローヌス,癲癇,小脳失調,筋緊張低下,知的退行,低身長
MTTK mitochondrial
DNA
筋緊張性ジストロフィー
刺激伝導系障害,心筋症,僧帽弁閉鎖不全筋強直,筋変性,白内障,眼瞼下垂
DMPK,ZNF9 19q13.2 3q13.3
Noonan症候群肺動脈狭窄,肥大型心筋症,心房中隔欠損翼状頚,低身長,発達遅滞,鳩胸,
漏斗胸,眼瞼下垂,出血傾向,血小板機能異常
PTPN11,KRAS,
SOS1,RAF1
12q24.1,
12p12.1,
2p22-p21,3p25
骨形成不全僧帽弁逸脱,大動脈弁閉鎖不全,大動脈拡大
骨脆弱性,多発骨折,難聴,青色強膜,下肢の彎曲変形および短縮,成長障害,顔貌異常
COL1A1
COL1A2
17q21.33
7q21.3
トリソミー13 心室中隔欠損,動脈管開存,心房中隔欠損
精神発達遅滞,全前脳胞症,小頭症,前額部傾斜,難聴,耳介変形,揺り椅子状足底,多指趾
Multiple Trisomy 13
Pompe病グリコーゲン蓄積による心筋肥大
先天性代謝異常,筋力低下,肝腫大,巨舌
GAA(Lysosomal Alpha-Glucosidase)
17q25
Rubinstein-Taybi 症候群
多岐の先天性心疾患,左心低形成発達障害,顔貌異常,多毛,眼瞼裂斜下,眼間開離,上顎低形
成,前額部突出,低身長,幅広い母指趾
CREBBP(CREB-binding protein)
16p13.3
Treacher-Collins 症候群
心室中隔欠損,動脈管開存,心房中隔欠損耳介形成異常,難聴,下顎形成不全,頬骨形成不全,脈絡膜欠損,両側下眼瞼部欠損,口蓋裂,後鼻腔閉鎖
TCOF1(Treacle protein)
5q32
結節性硬化症心臓腫瘍(横紋筋腫),不整脈腫瘍,痙攣,顔面血管線維種,
白斑,カフェオレ斑,骨硬化,腎形成異常,精神発達遅滞,自閉症
TSC1(Hamartin),TSC2(Tuberin)9q34
16p13.3
Turner症候群大動脈縮窄,大動脈二尖弁,左心低形成,心房中隔欠損,心室中隔欠損
低身長,翼状頚,盾状胸,毛髪線の低位,卵巣形成異常,腎形成異常,難聴
Multiple Unisomy X(45,X)
VACTERL連合心室中隔欠損,心房中隔欠損,動脈管開存脊柱の異常,鎖肛,気管食道瘻,
橈骨異形成,四肢の異常,腎泌尿器の異常
Numerous loci Unknown
22q11.2欠失症候群
大動脈離断,総動脈幹遺残,肺動脈閉鎖を伴うFallot四徴症,右側大動脈弓,鎖骨下動脈起始
異常,心室中隔欠損円錐動脈幹異常顔貌,鼻咽腔不全口蓋裂,胸腺低形成,副甲状腺機能低下,低カルシウム血症,易感染性,鎖肛,精神発達遅滞,精神障害,血小板減少症TBX1,UFD1L,
del 22q11.2
Williams 症候群大動脈弁上狭窄,肺動脈弁上狭窄,末梢性肺動脈狭窄,大動脈狭窄,肺動脈狭窄,心筋症
精神発達遅延,妖精様顔貌,虹彩の放射線模様,高カルシウム血症,不正咬合,視空間認知障害,関節拘縮,筋緊張亢進,学習障害,視覚認知障害ELN(Elastin) 7q11.23
疾患名心血管系異常心血管系以外の表現型疾患遺伝子,変異蛋白遺伝子座Alagille症候群末梢性肺動脈狭窄,肺動脈弁狭窄,Fallot四徴症,心室中隔欠損,心房中隔欠損,大動脈狭窄,大動脈縮窄
胆汁うっ滞,顔貌異常,精神発達遅滞,腎形成不全,眼異常,蝶型脊椎
JAG1(jagged-1)
NOTCH2
20p12
1p12
Barth症候群拡張型心筋症,左室緻密化障害神経筋異常,白血球減少,ミトコンドリア代謝異常,精神発達遅滞
TAZ(Tafazzin) Xq28
Cat-Eye症候群左心低形成,総肺脈還流異常,心室中隔欠損,心房中隔欠損虹彩裂,鎖肛,耳介変形,小顎症,腎形成異常
DGCR duplication
22q11.1
CHARGE連合Fallot四徴症, 房室中隔欠損,Ebstein病,完全大血管転位虹彩欠失,後鼻孔閉鎖,発達遅滞,腎形成異常,性器低形成,耳介変形,難聴,食道気管瘻
CHD7
SEMA3E
8q12.1
7q21.11
Down症候群房室中隔欠損,心室中隔欠損,心房中隔欠損,鎖骨下動脈起始異常顔貌異常,成長発達遅滞,十二
指腸閉鎖,鎖肛,気管軟化症,難聴,甲状腺機能低下,筋緊張低下,白血病
Multiple Trisomy 21
Duchenne型筋ジストロフィー心筋症,刺激伝導系障害,僧帽弁逸脱
進行性骨格筋萎縮DMD(Dystrophin) Xp21.2
トリソミー18 心室中隔欠損,動脈管開存,大動脈二尖弁,肺動脈二尖弁子宮内発育遅延,羊水過多,臍帯動脈異常,顔貌異常,精神運動発達遅滞,指の重なり,筋緊張低下
Multiple Trisomy 18
Ehlers-Danlos 症候群
僧帽弁逸脱,三尖弁逸脱 大動脈拡大,脳動脈瘤,心房中隔欠損皮膚の脆弱性,皮膚関節過伸展,皮下出血,青色強膜,気胸COL5A1,A2(Ⅰ,Ⅱ型)
COL3A1(Ⅳ型)
PLOD(Ⅳ型)
9q34.2-q34.3,
2q31
2q31
1p36.3
Ellis-Van Creveld症候群巨大心房中隔欠損,房室中隔欠損四肢短縮,多指症,爪低形成,骨盤異形成
EVC 4p16
Fabry病心筋虚血,心筋梗塞,僧帽弁閉鎖不全,左室肥大,心筋症,不整脈,うっ血性心不全四肢疼痛,知覚異常,被角血管腫,低汗症,腎不全,脳血管障害,角膜混濁,白内障,便秘,食道アカラシア,難聴
GAL(α-galactosidase) Xq22.1
Friedrich失調症心筋症,刺激伝導系障害進行性失調,筋緊張低下FRDA(Frataxin) 9q13
Goldenhar症候群心室中隔欠損, 動脈管開存,Fallot四徴症,大動脈縮窄,心房中隔欠損顔面非対称,脊椎異常,小耳症,下顎形成不全,難聴,眼球結膜類上皮腫
Unknown Unknown
内臓錯位症候群単心房,単心室,共通房室弁口,肺動脈閉鎖,大血管転位,房室中隔欠損,刺激伝導系障害
Kartagener症候群: 男性不妊,内臓逆位,気管支拡張症,難聴
ZIC3,LEFTY2,CFC1,
ACVR2B
Xq26.2,
3p22-p21.3,
Ivemark症候群:無脾症/多脾症1q42.1,2q21.1
Holt-Oram症候群心房中隔欠損,心室中隔欠損,刺激伝導系障害(洞性徐脈,房室ブロック)
橈骨系統異常(拇指異常,第2~第5指異常),上肢低形成
TBX5 12q24.1
ホモシステイン尿症
血栓塞栓症,大動脈拡大先天代謝異常,精神発達遅滞,骨格異常(高身長,四肢指伸長),水晶体偏位,精神障害,血栓塞
栓症,骨粗鬆症
MTHFR 1p36.3
Hurler症候群心筋症,房室弁および半月弁閉鎖不全先天代謝異常,特異的顔貌,進行性骨異形成,発達遅滞,角膜
混濁,難聴,成長障害,脊柱側彎,多毛症,肝脾腫
IDUA(Alpha-LIduronidase)
4p16.3
Jacobsen症候群左心低形成,心房中隔欠損,心室中隔欠損
精神運動発達遅滞,顔貌異常,足指関節の変形(槌趾症候群)
BARX2 Deletion 11q25
J e r v e l l - L a n g e -
Nielsen症候群QT 延長症候群難聴KCNQ1
KCNE1
11p15.5
21q22.1-q22.2
胆汁うっ滞,顔貌異常,精神発達遅滞,腎形成不全,眼異常,蝶型脊椎
JAG1(jagged-1)
NOTCH2
20p12
1p12
Barth症候群拡張型心筋症,左室緻密化障害神経筋異常,白血球減少,ミトコンドリア代謝異常,精神発達遅滞
TAZ(Tafazzin) Xq28
Cat-Eye症候群左心低形成,総肺脈還流異常,心室中隔欠損,心房中隔欠損虹彩裂,鎖肛,耳介変形,小顎症,腎形成異常
DGCR duplication
22q11.1
CHARGE連合Fallot四徴症, 房室中隔欠損,Ebstein病,完全大血管転位虹彩欠失,後鼻孔閉鎖,発達遅滞,腎形成異常,性器低形成,耳介変形,難聴,食道気管瘻
CHD7
SEMA3E
8q12.1
7q21.11
Down症候群房室中隔欠損,心室中隔欠損,心房中隔欠損,鎖骨下動脈起始異常顔貌異常,成長発達遅滞,十二
指腸閉鎖,鎖肛,気管軟化症,難聴,甲状腺機能低下,筋緊張低下,白血病
Multiple Trisomy 21
Duchenne型筋ジストロフィー心筋症,刺激伝導系障害,僧帽弁逸脱
進行性骨格筋萎縮DMD(Dystrophin) Xp21.2
トリソミー18 心室中隔欠損,動脈管開存,大動脈二尖弁,肺動脈二尖弁子宮内発育遅延,羊水過多,臍帯動脈異常,顔貌異常,精神運動発達遅滞,指の重なり,筋緊張低下
Multiple Trisomy 18
Ehlers-Danlos 症候群
僧帽弁逸脱,三尖弁逸脱 大動脈拡大,脳動脈瘤,心房中隔欠損皮膚の脆弱性,皮膚関節過伸展,皮下出血,青色強膜,気胸COL5A1,A2(Ⅰ,Ⅱ型)
COL3A1(Ⅳ型)
PLOD(Ⅳ型)
9q34.2-q34.3,
2q31
2q31
1p36.3
Ellis-Van Creveld症候群巨大心房中隔欠損,房室中隔欠損四肢短縮,多指症,爪低形成,骨盤異形成
EVC 4p16
Fabry病心筋虚血,心筋梗塞,僧帽弁閉鎖不全,左室肥大,心筋症,不整脈,うっ血性心不全四肢疼痛,知覚異常,被角血管腫,低汗症,腎不全,脳血管障害,角膜混濁,白内障,便秘,食道アカラシア,難聴
GAL(α-galactosidase) Xq22.1
Friedrich失調症心筋症,刺激伝導系障害進行性失調,筋緊張低下FRDA(Frataxin) 9q13
Goldenhar症候群心室中隔欠損, 動脈管開存,Fallot四徴症,大動脈縮窄,心房中隔欠損顔面非対称,脊椎異常,小耳症,下顎形成不全,難聴,眼球結膜類上皮腫
Unknown Unknown
内臓錯位症候群単心房,単心室,共通房室弁口,肺動脈閉鎖,大血管転位,房室中隔欠損,刺激伝導系障害
Kartagener症候群: 男性不妊,内臓逆位,気管支拡張症,難聴
ZIC3,LEFTY2,CFC1,
ACVR2B
Xq26.2,
3p22-p21.3,
Ivemark症候群:無脾症/多脾症1q42.1,2q21.1
Holt-Oram症候群心房中隔欠損,心室中隔欠損,刺激伝導系障害(洞性徐脈,房室ブロック)
橈骨系統異常(拇指異常,第2~第5指異常),上肢低形成
TBX5 12q24.1
ホモシステイン尿症
血栓塞栓症,大動脈拡大先天代謝異常,精神発達遅滞,骨格異常(高身長,四肢指伸長),水晶体偏位,精神障害,血栓塞
栓症,骨粗鬆症
MTHFR 1p36.3
Hurler症候群心筋症,房室弁および半月弁閉鎖不全先天代謝異常,特異的顔貌,進行性骨異形成,発達遅滞,角膜
混濁,難聴,成長障害,脊柱側彎,多毛症,肝脾腫
IDUA(Alpha-LIduronidase)
4p16.3
Jacobsen症候群左心低形成,心房中隔欠損,心室中隔欠損
精神運動発達遅滞,顔貌異常,足指関節の変形(槌趾症候群)
BARX2 Deletion 11q25
J e r v e l l - L a n g e -
Nielsen症候群QT 延長症候群難聴KCNQ1
KCNE1
11p15.5
21q22.1-q22.2
5 遺伝カウンセリング
① 総論
心疾患患者の妊娠を考える際には,心疾患の親子間の繰り返しの可能性など,遺伝学的な知識を正確に伝える必要がある.その意味において,心疾患患者の妊娠・出産に関連した,遺伝カウンセリングや遺伝相談の重要性は増している.一方で,遺伝カウンセリングを行うにあたっては,対象者本人の自己決定権,わかりやすい十分な説明,未成年者への配慮,得られた情報の守秘義務など,基本的人権に関与する事態への慎重な対応が強く求められる.
遺伝カウンセリングでは,相談に来る人をクライエント,相談に応じる人をカウンセラーと呼ぶ.遺伝カウンセリングは,習熟した専門スタッフ(臨床遺伝専門医が望ましい)がコメディカルスタッフ(遺伝カウンセラー)と共同して行う.カウンセリングの内容が,単なる遺伝医学情報の提供にとどまらず,クライエントの立場に立った,問題解決の援助,心理的な対応をする技術,高度な倫理的内容と知識なども必要とされるようになり,臨床遺伝専門医とは別に,専門職の遺伝カウンセラーの果たす役割が大きくなった.遺伝カウンセラーは,医師が診断を下すことを補助する,再発リスクを評価する,再発に関する情報を患者がわかる言葉で説明する,患者が適切な決断を下して行動がとれるように手助けをする,などの仕事を担当する.
遺伝カウンセリングにおいてはいくつかの重要な注意点がある.遺伝医学関連10学会および研究会の提唱する「遺伝学的検査に関するガイドライン」69)によると,「遺伝カウンセリングは,遺伝性疾患の患者・家族またはその可能性のある人(クライエント)に対して,生活設計上の選択を自らの意志で決定し行動できるよう臨床遺伝学診断を行い,遺伝医学的判断に基づき遺伝予後等の適切な情報を提供し,支援する医療行為である」とされ,「遺伝カウンセリングにおいては,クライエントと遺
伝カウンセリングの担当者との良好な信頼関係に基づき,様々なコミュニケーションが行われ,その過程で心理的精神的援助がなされる.遺伝カウンセリングは決して一方的な遺伝医学的情報提供だけではないことに留意すべきである」と記載されている.すなわち,遺伝カウンセリングは,あくまでもクライエントの自由意志による相談であり,カウンセラーが遺伝的側面に関するあらゆる情報を提供することにより,それを参考にしてクライエントが自己決定することが重要である.この決定は他人から強制されてはならない.また遺伝カウンセリングにおいては,クライエントの人権と利益の尊重を最優先課題とし,また得られたクライエント個人の遺伝情報を厳密
に守秘する必要がある.
遺伝カウンセリングの原則は,遺伝関連10学会および研究会が提案した「遺伝学的検査に関するガイドライン(2003)」69)に,また,心疾患系疾患における遺伝カウ
ンセリングの様々な留意点は,日本循環器学会の「心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン(2006)」70)に詳細が記載されている.以下に「遺伝学的検査に関するガイドライン(2003)」より遺伝学的検査と遺伝カウンセリングの項目を抜粋して掲載する.
② 遺伝学的検査と遺伝カウンセリング(「遺伝学的検査に関するガイドライン(2003)」69)より)
1)遺伝学的検査と遺伝カウンセリングの大原則
(1)遺伝学的検査は,十分な遺伝カウンセリングを行った後に実施する.
(2)遺伝カウンセリングは,十分な遺伝医学的知識・経験を持ち,遺伝カウンセリングに習熟した臨床遺伝専門医などにより,被検者の心理状態を常に把握しながら
行う必要がある.遺伝カウンセリング担当者は,必要に応じて,精神科医,臨床心理専門職,遺伝看護師,ソーシャルワーカーなどの協力を求め,チームで行う
ことが望ましい.
(3)遺伝カウンセリング担当者は,できる限り正確で最新の関連情報を被検者に提供するように努める.これには疾患の頻度,自然歴,再発率(遺伝的予後),
さらに保因者検査,出生前検査,発症前検査,易罹患性検査などの遺伝学的検査の意味についての情報が含まれる.遺伝カウンセリング担当者は,遺伝性
疾患が同一疾患であっても,その遺伝子変異,臨床像,予後,治療効果などにおいて異質性に富むことが多いことについて,十分留意する必要がある.
(4)遺伝カウンセリング担当者は,被検者が理解できる平易な言葉を用い,被検者が十分理解していることを常に確認しながら,遺伝カウンセリングを進める必要が
ある.被検者の依頼がある場合,またはその必要があると判断される場合は,被検者以外の人物の同席を考慮する.
(5)遺伝カウンセリングの内容は,一般診療録とは別の遺伝カウンセリング記録簿に記載し,一定期間保存する.
(6)被検者が望んだ場合,被検者が自由意志で決定できるように,遺伝カウンセリングは継続して行うことが望ましい.また必要に応じて,臨床心理的,社会的支援
を含めた,医療・福祉面での対応について,情報提供する必要がある.
(7)遺伝学的診断結果が,担当医師によって,被検者の血縁者にも開示されるような場合には,臨床遺伝専門医の紹介など,その血縁者も遺伝カウンセリングを受
けられるように配慮する.
(8)遺伝カウンセリングは,遺伝学的検査の実施後も,必要に応じて行う.
2)目的に応じた遺伝学的検査における留意点
①発症者を対象とする遺伝学的検査
(1)遺伝学的検査は,発症者の確定診断を目的として行われることがある.
(2)発症者の確定診断の目的で行われる遺伝学的検査の場合も,結果的にその情報が,血縁者に影響を与える可能性があることについて,検査前に十分説明し,
理解を得ておく必要がある.
(3)血縁者の発症前診断,易罹患性診断,保因者診断などを行うための情報を得ることを第一の目的として,既に臨床診断が確定している患者に対して,疾患の原
因となっている遺伝子変異などを解析することがある.この場合は,得られた情報が適切に血縁者に開示されるか,あるいは利用されることによって初めて意味
のある遺伝学的検査となること,疾患の原因となる遺伝子変異が見出されなくても,本人の臨床診断に影響しないことを,検査の前に被検者に十分説明し,理
解を得ておく必要がある.
②保因者の判定を目的とする遺伝学的検査
(1)遺伝学的検査は,家系内に常染色体劣性遺伝病やX連鎖劣性遺伝病,染色体不均衡型構造異常の患者がいる場合,当事者が保因者であるかどうかを明ら
かにし,将来,子孫が同じ遺伝病に罹患する可能性を予測するための保因者検査として行われることがある.
(2)保因者検査を行うにあたっては,被検者に対して,その検査が直接本人の健康管理に役立つ情報を得る目的のものではなく,将来の生殖行動に役立つ可能
性のある情報を得るために行われるものであることを十分に説明し,理解を得る必要がある.
(3)将来の自由意思の保護という観点から,小児に対する保因者診断は基本的に行わないことが望ましい.
(4)保因者検査を行う場合には,担当医師および関係者は,診断の結果明らかになる遺伝的特徴に基づいて,被検者およびその血縁者ならびに家族が社会的な
差別を受ける可能性について十分に配慮する.
③発症予測を目的とする遺伝学的検査
(1)発症を予測する遺伝学的検査には,単一遺伝子の変異でほぼ完全に発症を予測することのできる発症前検査と,多因子疾患の罹患性の程度もしくは罹病リ
スクを予測する易罹患性検査がある.
(2)発症予測を目的とする遺伝学的検査の対象者は,一般に健常者であるため,厳格なプライバシーの保護および適切な心理的援助が措置される必要がある.
特に就学,雇用および昇進,ならびに保険加入などに際して,差別を受けることのないように配慮する.
a.発症前検査
(1)有効な治療法および予防法の確立されていない疾患の発症前検査においては,以下のすべての要件が満たされない限り,行わない.
◦被検者は判断能力のある成人であり,被検者が自発的に発症前検査を希望していること.
◦同一家系内の罹患者の遺伝子変異が判明しているなど,遺伝学的検査によって確実に診断できること.
◦被検者は当該疾患の遺伝形式,臨床的特徴,遺伝学的検査法の詳細についてよく理解しており,検査の結果が陽性であった場合の将来設計について熟慮し
いること.
◦遺伝学的検査後,結果が陽性であった場合には,発症後においても,臨床心理的・社会的支援を含むケアおよび治療を行う医療機関が利用できること.
(2)有効な治療法および予防法が確立されていない疾患の発症前検査は,前項の要件がすべて満たされている場合に限り,かつ当該疾患の専門医・臨床遺伝専
門医などを含む複数の医師により,可能な限り,臨床心理専門職・看護師・ソーシャルワーカーなどの協力を得て,複数回の遺伝カウンセリングを行った上で,
実施の可否を慎重に決定する.
b.易罹患性検査
(1)多因子疾患などに関する易罹患性検査を行う場合には,検査の感度,特異度,陽性・陰性結果の正診率などが十分なレベルにあることを確認する.
(2)易罹患性検査に際しては,担当医師は,遺伝子(DNA)変異が同定されても,その発症は疾患により一様ではなく浸透率や罹患性に対する効果(寄与率)など
に依存すること,また,検査目標とする遺伝子に変異が見出されない場合であっても発症する可能性が否定できないことなどについて,被検者に十分に説明し,
理解を求める.
c.家族性腫瘍に関する検査
(1)易罹患性検査のうち,家族性腫瘍に関する検査に関しては,関連遺伝子の多様性に配慮した,慎重な対応をする.
(2)家族性腫瘍の易罹患性検査に関しては,本ガイドラインに加えて,家族性腫瘍研究会の「家族性腫瘍における遺伝子診断の研究とこれを応用した診療に関する
ガイドライン」に準拠する.
(3)家族性腫瘍の易罹患性検査を行うにあたっては,検査の感度,特異度,陽性・陰性結果の正診率などが十分なレベルにあることを確認する.
④薬物に対する反応性の個体差を判定することを目的とする遺伝学的検査
薬物代謝酵素の遺伝子多型検査による薬剤感受性診断は,直接治療に役立て得る情報であり,有用性が高いと考えられるが,この情報が社会的差別などに誤用されることのないよう,他の目的の遺伝学的検査と同様の注意が必要である.
⑤出生前検査と出生前診断
(1)妊娠前半期に行なわれる出生前検査および診断には,羊水,絨毛,その他の胎児試料などを用いた細胞遺伝学的,遺伝生化学的,分子遺伝学的,細胞・病理
学的方法,および超音波検査などを用いた物理学的方法などがある.
(2)出生前検査および診断として遺伝学的検査および診断を行うにあたっては,倫理的および社会的問題を包含していることに留意し,特に以下の点に注意して実
施する.
◦胎児が罹患児である可能性(リスク),検査法の診断限界,母体・胎児に対する危険性,副作用などについて検査前によく説明し,十分な遺伝カウンセリングを
行うこと.
◦検査の実施は,十分な基礎的研修を行い,安全かつ確実な検査技術を習得した産科医により,またはその指導の下に行われること.
(3)絨毛採取,羊水穿刺など,侵襲的な出生前検査・診断は,下記のような場合の妊娠について夫婦からの希望があり,検査の意義について十分な理解が得られ
た場合に行う.
◦夫婦のいずれかが,染色体異常の保因者である場合 .
◦染色体異常症に罹患した児を妊娠,分娩した既往を有する場合.
◦高齢妊娠の場合 .
◦妊婦が新生児期もしくは小児期に発症する重篤なX連鎖遺伝病のヘテロ接合体の場合 .
◦夫婦のいずれもが,新生児期もしくは小児期に発症する重篤な常染色体劣性遺伝病のヘテロ接合体の場合 .
◦夫婦のいずれかが,新生児期もしくは小児期に発症する重篤な常染色体優性遺伝病のヘテロ接合体の場合 .
◦その他,胎児が重篤な疾患に罹患する可能性のある場合.
(4)重篤なX連鎖遺伝病のために検査が行われる場合を除き,胎児の性別を告げない.
(5)出生前診断技術の精度管理については,常にその向上に努める.
(6)母体血清マーカー検査の取り扱いに関しては,厚生科学審議会先端医療技術評価部会出生前診断に関する専門委員会による「母体血清マーカー検査に関
する見解」,日本人類遺伝学会倫理審議委員会による「母体血清マーカー検査に関する見解」,および日本産科婦人科学会周産期委員会による報告「母体血
清マーカー検査に関する見解について」を十分に尊重して施行する.
(7)着床前検査および診断は極めて高度な知識・技術を要する,いまだ研究段階にある遺伝学的検査を用いた医療技術であり,倫理的側面からもより慎重に取り
扱う.実施に際しては,日本産科婦人科学会会告「着床前診断に関する見解」に準拠する.
③ 先天性心血管疾患
1)遺伝的要因
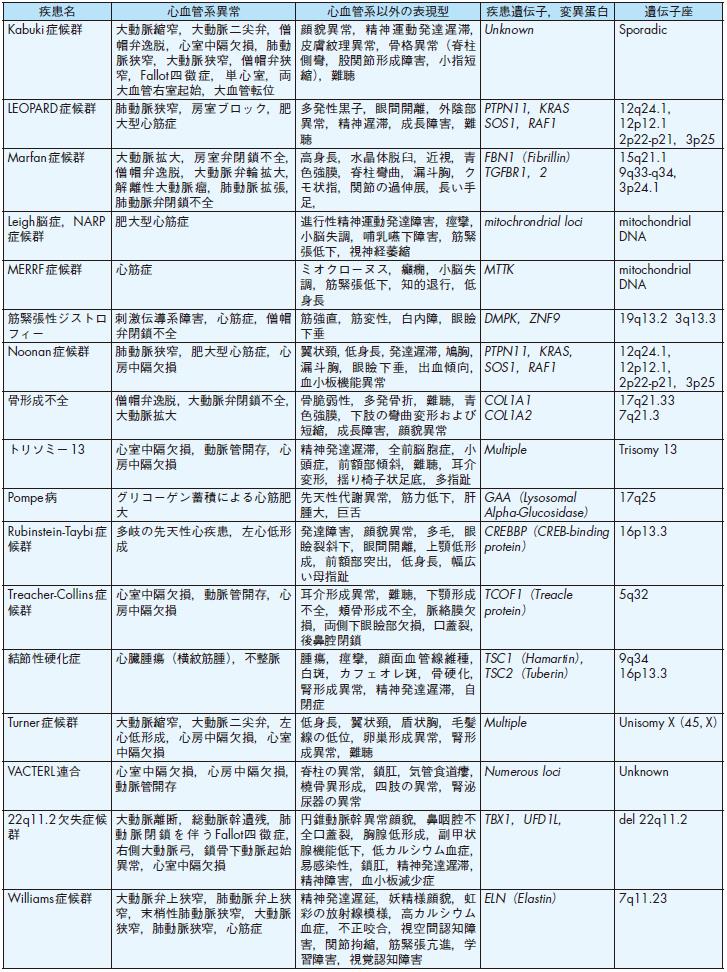
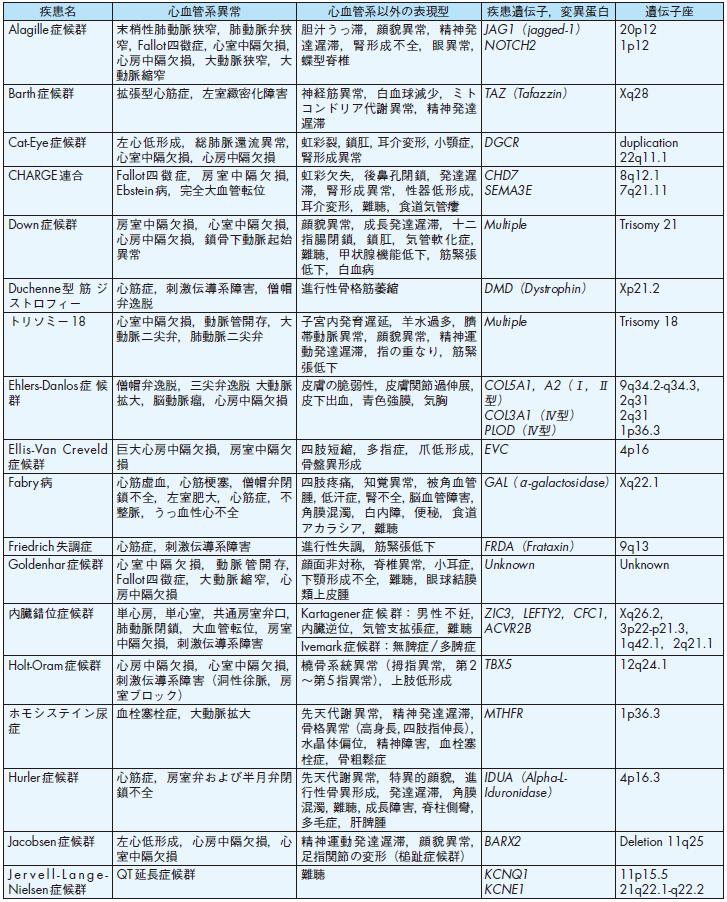
旧厚生省班研究の全国調査(1986年)71)では,日本における先天性心血管疾患の発生頻度は生産児の1.06%であり,新生児死亡の原因となる先天性疾患の中では最も頻度が高い.一方,日本小児循環器学会疫学委員会の調査(2003年)72)では,その成因による内訳は,染色体異常(Down症候群,Turner 症候群,22q11.2欠失症候群,Williams症候群など,8.2%)や遺伝子病(Noonan症候群,Holt-Oram症候群,Marfan症候群,遺伝性QT延長症候群など,4.7%)などの遺伝要因によるものが全体の約12.9%,母体の全身疾患,胎内感染,催奇形因子など,環境(外的)要因によるものが0.5%であり,残りの86.7%は成因不明の多因子遺伝(多くの先天性心疾患,特発性肺高血圧症,特発性心筋症など)によると報告されている.多因子遺伝とは,個人の遺伝的素因(遺伝子異常)と環境(外的)因子との相互関係によって疾患が発生するものを指す.すなわち,純粋にメンデル遺伝のみで説明できる先天性心血管疾患は一部であり,大半は胎内での形態形成,生後の発達・発育,加齢に伴う環境要因によって疾患遺伝子の関与の程度が左右され,表現形の多様性と深くかかわっていることが明らかになってきた73)-76).一方,近年のゲノム医学ならび遺伝子改変マウスを用いた発生工学の目覚ましい発展により,多くの遺伝性先天性心血管疾患の責任遺伝子が明らかになってきた.その代表的な疾患と表現型および疾患遺伝子を表5に掲載する73)-75),77),78).各疾患の臨床像や妊娠・出産における問題点に関しては,本ガイドライン後半の各論の項を参照されたい.また,先天性心血管疾患における遺伝的要因に関する詳細は,American Heart Association Congenital Cardiac Defects Committeeより出された声明78),およびボストン小児病院より出されているホームページCongenital Cardiovascular Genetics Program 77)を参照されたい.
2)環境要因
また先天性心血管疾患の成因には,遺伝子異常のみならず,妊娠中の胎児および母体の環境要因も重要である79).妊婦の喫煙は児の心室中隔欠損症,Fallot四徴症,心房中隔欠損症,完全大血管転位症の発症の危険因子であることが,また妊婦の飲酒は中枢神経系の障害を伴う胎児性アルコール症候群とともに,Fallot四徴症,心房中隔欠損症,完全大血管転位症の発症と関連することが報告されている72),80).また,母体が妊娠早期に風疹ウイルスに感染すると,先天性風疹症候群として,先天性白内障や先天性難聴とともに心室中隔欠損症,心房中隔欠損症,肺動脈狭窄症が高率に発症する79).血糖値のコントロールが不良な糖尿病の母体からは,肥大型心筋症,内臓錯位症候群,房室中隔欠損症,完全大血管転位症などが発症する79).またSLEやシェーグレン症候群に罹患した妊婦では,抗SSA,抗SSB抗体が経胎盤的に移行して児に高度房室ブロックが発症することがあり,治療として1度房室ブロック段階での母体へのステロイド投与が推奨されている81),82).フェニルケトン尿症では,高フェニルアラニン血症によりFallot四徴症,心室中隔欠損症,心房中隔欠損症,大動脈縮窄症,左心低形成症候群などが発症する.妊娠前からの低フェニルアラニン食によるコントロールが必要となる83).また,胎児に先天性心血管疾患をもたらす可能性のある薬剤として,アンフェタミン(心室中隔欠損症,動脈管開存症,心房中隔欠損症など),ヒダントイン(肺動脈狭窄症,大動脈狭窄症,大動脈縮窄症など),トリメタジオン(完全大血管転位症,Fallot四徴症,左心低形成症候群など),リチウム(Ebstein病,三尖弁閉鎖症など)が報告されている73)-75),79).妊婦が摂取するビタミンの過不足も先天性心血管異常と深い関係がある.ビタミンAの過剰摂取は胎児に,心臓流出路障害とともに中枢神経系や口蓋や胸腺の異常を来たす76),84).妊婦の葉酸摂取が不足すると胎児に心室中隔欠損症などの心血管異常を来たすため,食品への葉酸の添加もなされている76),79),85),86).母体の有機溶媒への曝露も胎児に房室中隔欠損症,完全大血管転位症,肺動脈狭窄症など様々な先天性心疾患をもたらす79).
3)各疾患群にみられる遺伝子異常
①先天性心疾患
先天性心疾患の多くは多因子遺伝と考えられており,メンデル遺伝様式には従わないことの方が多い.しかしながら,先天性心疾患を持つ親から生まれる子供たちは先天性心疾患を繰り返す頻度が高く,一般頻度に比べて3〜5倍と考えられている72),74),76).この場合,母親が先天性心疾患の場合,父親が先天性心疾患である場合より頻度は2倍以上高い.日本小児循環器学会の全国調査によると,親子間での疾患一致率は,心房中隔欠損症(66.7%)および動脈管開存症(66.7%)が最も高く,次いで心室中隔欠損症(47.1%),Fallot四徴症(28.6%)である72).一方,最近の発生生物学ならび遺伝子工学の目覚ましい発展により,先天性心疾患の原因となる数多くの遺伝子異常が明らかになってきた.前述の表5に原因が明らかになった代表的な遺伝性先天性心血管疾患を示した7),9).
②特発性心筋症
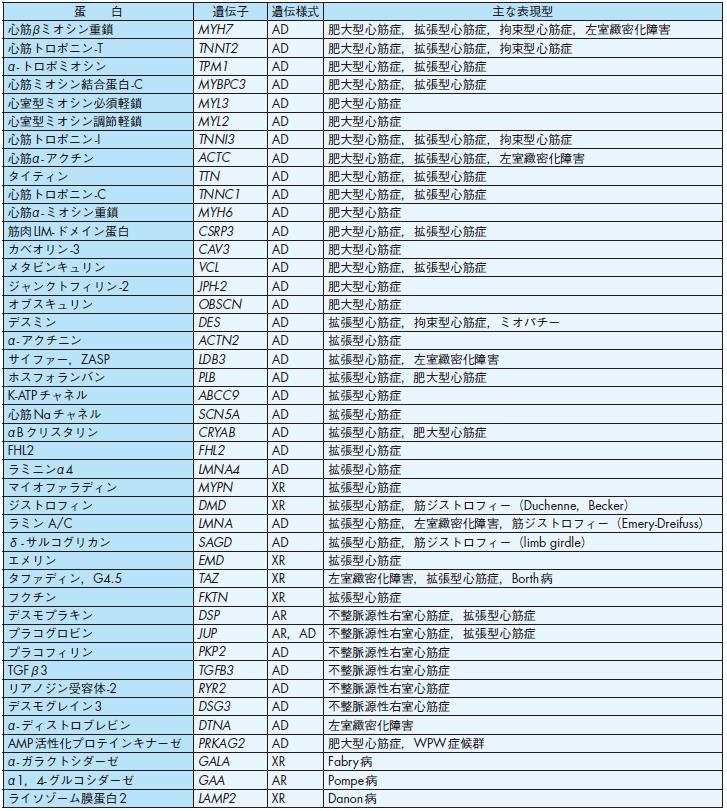
肥大型心筋症の約50%に常染色体性優性遺伝による家族歴が認められる87).ミオシン,アクチン,トロポニン,トロポミオシンなどのサルコメア収縮に直接関連す
る要素以外にも,タイチンなどの細胞骨格や膜蛋白カベオリンなどのシグナル伝達物質の異常でも肥大型心筋症が発症することが明らかとなり87),88)(表6),筋細胞肥大の原因として筋収縮におけるカルシウム感受性の亢進が考えられている89).家族性肥大型心筋症では,約50%に遺伝子異常が明らかにされている.その中では,心筋βミオシン重鎖(MHY7)での変異が約20%,心筋トロポニンT(TNNT2)および心筋ミオシン結合蛋白(MYBPC3)での変異が各々約10%であり,これらの3
種類の家族性肥大型心筋症の主要な原因遺伝子となっている.その中では,MYH7 およびMYBPC3の変異例では肥大の程度が強く,TNNT2 の変異例では肥大の程度は軽いが心収縮力の低下し比較的早期より拡張相に移行する.弧発例の肥大型心筋症おいても約15%に変異が見つかる87),88)(レベルC).
肥大型心筋症類似の病態を示す疾患として,多くの蓄積病の原因遺伝子も明らかになってきた.Pompe病(α1,4- グルコシダーゼ欠損),Danon病(リソゾーム膜蛋
白欠損症),Fabry病(αガラクトシダーゼA欠損症),AMP活性化蛋白キナーゼ変異症などでは心肥大がみられる87),88),90).
拡張型心筋症の家族内発症は肥大型心筋症に比べて少なく,約30%とされている.原因遺伝子としては,αアクチニン,デスミン,タイチン,ジストロフィンなど,心筋細胞骨格や細胞接着分子に変異が認められることが多いが,肥大型心筋症同様にサルコメア変異も報告されている87),88),91) (表6).不整脈原性右室心筋症は,臨床的に右心室に期限を有する心室性不整脈を主症状とし,病理組織学的に右心室優位の心筋細胞の変性,脱落と線維脂肪浸潤の増加を示す疾患である.遺伝子異常として,約30%の症例にプラコフィリン2(PKP2)が,またその他に,デスモプラキン,プラコグロビン,プラコフィリン,リアノジン受容体-2,TGFβ 3などの遺伝子変異が報告されている92),93)(レベルC).
ミトコンドリア心筋症は,ミトコンドリアにおけるエネルギー産生障害に基づいて引き起こされるミトコンドリア病の中で,肥大型心筋症,拡張型心筋症,拘束型心筋症,刺激伝導障害などの多様な心病変を示す代謝性心筋症である95),96).一般にミトコンドリア病では,中枢神経障害(精神運動発達遅滞,痙攣,意識消失),筋力低下,乳酸アシドーシスなどの全身症状がみられることがある.ミトコンドリア心筋症は母系遺伝を示すことが多く,無症状の母方家族に変異が検出されることがあり,遺伝学的な配慮が必要である.
これらすべての遺伝性心筋疾患においては,妊娠に伴う循環動態の変化により生じる母体および児へのリスクを十分に説明するとともに,児への疾患の繰り返しに関する遺伝的情報を説明する必要がある.
③遺伝性不整脈
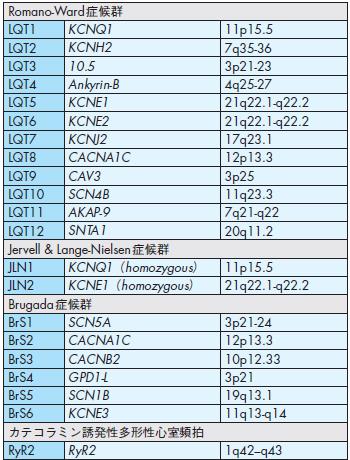
突然死の原因となる遺伝性QT延長症候群,カテコラミン誘発多形性心室頻拍,Brugada症候群,進行性心臓伝導障害,家族性洞機能不全症候群,家族性心房細動,QT短縮症候群などにおいて,遺伝子変異が明らかとなっている96)-98)(表7).
QT延長症候群は,基礎疾患がみられずに心電図上QT間隔が延長し,心室頻拍や心室細動から失神および突然死に至る可能性のある疾患群である.現在までに,カリウムおよびナトリウムチャネル遺伝子を中心として,LQT1-LQT12まで分類されており,300以上の遺伝子異常が報告されている97)-99).その内訳は,LQT1(約40%),LQT2(約30〜40%),LQT3(約10%)とされており,LQT1〜LQT3で全体の約90%を占める.一般にLQT1は運動中や水泳中に,LQT2では睡眠中の聴覚刺激(目覚まし時計など)により,LQT3 では安静時および睡眠中に心事故を起こす.
Brugada症候群は,右側胸部誘導心電図(V1-V3)にて右脚ブロック様の心電図波形と特徴的なST上昇(coved 型またはsaddle back型)を伴う,中年男性に多い特発性心室細動の症候群である.責任遺伝子としてナトリウムチャネルαサブユニット(SCN5A)の異常が15〜25%に報告されている100).
カテコラミン誘発多形性心室頻拍は,学童や青年期において運動や交感神経の刺激に伴い心室頻拍から失神や突然死を来たす症候群であり,心筋細胞小胞体のリアノジン受容体(RyR2)の遺伝子異常が報告されている101).
遺伝性不整脈疾患の内服治療の詳細および植込み型除細動器の適応については,日本循環器学会「QT延長症候群(先天性・二次性)とBrugada症候群の診療に関するガイドライン(2007年)」102)を参照されたい.
これらすべての遺伝性不整脈疾患においても,妊娠に伴う母体および児へのリスクを十分に説明するとともに,児への疾患の繰り返しに関する遺伝的情報をカウン
セリングする必要がある.
④その他の先天性心血管疾患
22q11.2欠失症候群,Marfan症候群,Loeys-Dietz症候群,Ehlers-Danlos 症候群,原発性肺高血圧症,Eisenmenger症候群,先天性心疾患術後遺残病変,Fontan手術後などにおいては,妊娠・出産に際して生じる母体の循環動態の変化に伴い,母親と胎児の厳重な管理が必要である.また,児への疾患の繰り返しに関する遺伝的情報を説明する必要がある.
心疾患患者の妊娠を考える際には,心疾患の親子間の繰り返しの可能性など,遺伝学的な知識を正確に伝える必要がある.その意味において,心疾患患者の妊娠・出産に関連した,遺伝カウンセリングや遺伝相談の重要性は増している.一方で,遺伝カウンセリングを行うにあたっては,対象者本人の自己決定権,わかりやすい十分な説明,未成年者への配慮,得られた情報の守秘義務など,基本的人権に関与する事態への慎重な対応が強く求められる.
遺伝カウンセリングでは,相談に来る人をクライエント,相談に応じる人をカウンセラーと呼ぶ.遺伝カウンセリングは,習熟した専門スタッフ(臨床遺伝専門医が望ましい)がコメディカルスタッフ(遺伝カウンセラー)と共同して行う.カウンセリングの内容が,単なる遺伝医学情報の提供にとどまらず,クライエントの立場に立った,問題解決の援助,心理的な対応をする技術,高度な倫理的内容と知識なども必要とされるようになり,臨床遺伝専門医とは別に,専門職の遺伝カウンセラーの果たす役割が大きくなった.遺伝カウンセラーは,医師が診断を下すことを補助する,再発リスクを評価する,再発に関する情報を患者がわかる言葉で説明する,患者が適切な決断を下して行動がとれるように手助けをする,などの仕事を担当する.
遺伝カウンセリングにおいてはいくつかの重要な注意点がある.遺伝医学関連10学会および研究会の提唱する「遺伝学的検査に関するガイドライン」69)によると,「遺伝カウンセリングは,遺伝性疾患の患者・家族またはその可能性のある人(クライエント)に対して,生活設計上の選択を自らの意志で決定し行動できるよう臨床遺伝学診断を行い,遺伝医学的判断に基づき遺伝予後等の適切な情報を提供し,支援する医療行為である」とされ,「遺伝カウンセリングにおいては,クライエントと遺
伝カウンセリングの担当者との良好な信頼関係に基づき,様々なコミュニケーションが行われ,その過程で心理的精神的援助がなされる.遺伝カウンセリングは決して一方的な遺伝医学的情報提供だけではないことに留意すべきである」と記載されている.すなわち,遺伝カウンセリングは,あくまでもクライエントの自由意志による相談であり,カウンセラーが遺伝的側面に関するあらゆる情報を提供することにより,それを参考にしてクライエントが自己決定することが重要である.この決定は他人から強制されてはならない.また遺伝カウンセリングにおいては,クライエントの人権と利益の尊重を最優先課題とし,また得られたクライエント個人の遺伝情報を厳密
に守秘する必要がある.
遺伝カウンセリングの原則は,遺伝関連10学会および研究会が提案した「遺伝学的検査に関するガイドライン(2003)」69)に,また,心疾患系疾患における遺伝カウ
ンセリングの様々な留意点は,日本循環器学会の「心臓血管疾患における遺伝学的検査と遺伝カウンセリングに関するガイドライン(2006)」70)に詳細が記載されている.以下に「遺伝学的検査に関するガイドライン(2003)」より遺伝学的検査と遺伝カウンセリングの項目を抜粋して掲載する.
② 遺伝学的検査と遺伝カウンセリング(「遺伝学的検査に関するガイドライン(2003)」69)より)
1)遺伝学的検査と遺伝カウンセリングの大原則
(1)遺伝学的検査は,十分な遺伝カウンセリングを行った後に実施する.
(2)遺伝カウンセリングは,十分な遺伝医学的知識・経験を持ち,遺伝カウンセリングに習熟した臨床遺伝専門医などにより,被検者の心理状態を常に把握しながら
行う必要がある.遺伝カウンセリング担当者は,必要に応じて,精神科医,臨床心理専門職,遺伝看護師,ソーシャルワーカーなどの協力を求め,チームで行う
ことが望ましい.
(3)遺伝カウンセリング担当者は,できる限り正確で最新の関連情報を被検者に提供するように努める.これには疾患の頻度,自然歴,再発率(遺伝的予後),
さらに保因者検査,出生前検査,発症前検査,易罹患性検査などの遺伝学的検査の意味についての情報が含まれる.遺伝カウンセリング担当者は,遺伝性
疾患が同一疾患であっても,その遺伝子変異,臨床像,予後,治療効果などにおいて異質性に富むことが多いことについて,十分留意する必要がある.
(4)遺伝カウンセリング担当者は,被検者が理解できる平易な言葉を用い,被検者が十分理解していることを常に確認しながら,遺伝カウンセリングを進める必要が
ある.被検者の依頼がある場合,またはその必要があると判断される場合は,被検者以外の人物の同席を考慮する.
(5)遺伝カウンセリングの内容は,一般診療録とは別の遺伝カウンセリング記録簿に記載し,一定期間保存する.
(6)被検者が望んだ場合,被検者が自由意志で決定できるように,遺伝カウンセリングは継続して行うことが望ましい.また必要に応じて,臨床心理的,社会的支援
を含めた,医療・福祉面での対応について,情報提供する必要がある.
(7)遺伝学的診断結果が,担当医師によって,被検者の血縁者にも開示されるような場合には,臨床遺伝専門医の紹介など,その血縁者も遺伝カウンセリングを受
けられるように配慮する.
(8)遺伝カウンセリングは,遺伝学的検査の実施後も,必要に応じて行う.
2)目的に応じた遺伝学的検査における留意点
①発症者を対象とする遺伝学的検査
(1)遺伝学的検査は,発症者の確定診断を目的として行われることがある.
(2)発症者の確定診断の目的で行われる遺伝学的検査の場合も,結果的にその情報が,血縁者に影響を与える可能性があることについて,検査前に十分説明し,
理解を得ておく必要がある.
(3)血縁者の発症前診断,易罹患性診断,保因者診断などを行うための情報を得ることを第一の目的として,既に臨床診断が確定している患者に対して,疾患の原
因となっている遺伝子変異などを解析することがある.この場合は,得られた情報が適切に血縁者に開示されるか,あるいは利用されることによって初めて意味
のある遺伝学的検査となること,疾患の原因となる遺伝子変異が見出されなくても,本人の臨床診断に影響しないことを,検査の前に被検者に十分説明し,理
解を得ておく必要がある.
②保因者の判定を目的とする遺伝学的検査
(1)遺伝学的検査は,家系内に常染色体劣性遺伝病やX連鎖劣性遺伝病,染色体不均衡型構造異常の患者がいる場合,当事者が保因者であるかどうかを明ら
かにし,将来,子孫が同じ遺伝病に罹患する可能性を予測するための保因者検査として行われることがある.
(2)保因者検査を行うにあたっては,被検者に対して,その検査が直接本人の健康管理に役立つ情報を得る目的のものではなく,将来の生殖行動に役立つ可能
性のある情報を得るために行われるものであることを十分に説明し,理解を得る必要がある.
(3)将来の自由意思の保護という観点から,小児に対する保因者診断は基本的に行わないことが望ましい.
(4)保因者検査を行う場合には,担当医師および関係者は,診断の結果明らかになる遺伝的特徴に基づいて,被検者およびその血縁者ならびに家族が社会的な
差別を受ける可能性について十分に配慮する.
③発症予測を目的とする遺伝学的検査
(1)発症を予測する遺伝学的検査には,単一遺伝子の変異でほぼ完全に発症を予測することのできる発症前検査と,多因子疾患の罹患性の程度もしくは罹病リ
スクを予測する易罹患性検査がある.
(2)発症予測を目的とする遺伝学的検査の対象者は,一般に健常者であるため,厳格なプライバシーの保護および適切な心理的援助が措置される必要がある.
特に就学,雇用および昇進,ならびに保険加入などに際して,差別を受けることのないように配慮する.
a.発症前検査
(1)有効な治療法および予防法の確立されていない疾患の発症前検査においては,以下のすべての要件が満たされない限り,行わない.
◦被検者は判断能力のある成人であり,被検者が自発的に発症前検査を希望していること.
◦同一家系内の罹患者の遺伝子変異が判明しているなど,遺伝学的検査によって確実に診断できること.
◦被検者は当該疾患の遺伝形式,臨床的特徴,遺伝学的検査法の詳細についてよく理解しており,検査の結果が陽性であった場合の将来設計について熟慮し
いること.
◦遺伝学的検査後,結果が陽性であった場合には,発症後においても,臨床心理的・社会的支援を含むケアおよび治療を行う医療機関が利用できること.
(2)有効な治療法および予防法が確立されていない疾患の発症前検査は,前項の要件がすべて満たされている場合に限り,かつ当該疾患の専門医・臨床遺伝専
門医などを含む複数の医師により,可能な限り,臨床心理専門職・看護師・ソーシャルワーカーなどの協力を得て,複数回の遺伝カウンセリングを行った上で,
実施の可否を慎重に決定する.
b.易罹患性検査
(1)多因子疾患などに関する易罹患性検査を行う場合には,検査の感度,特異度,陽性・陰性結果の正診率などが十分なレベルにあることを確認する.
(2)易罹患性検査に際しては,担当医師は,遺伝子(DNA)変異が同定されても,その発症は疾患により一様ではなく浸透率や罹患性に対する効果(寄与率)など
に依存すること,また,検査目標とする遺伝子に変異が見出されない場合であっても発症する可能性が否定できないことなどについて,被検者に十分に説明し,
理解を求める.
c.家族性腫瘍に関する検査
(1)易罹患性検査のうち,家族性腫瘍に関する検査に関しては,関連遺伝子の多様性に配慮した,慎重な対応をする.
(2)家族性腫瘍の易罹患性検査に関しては,本ガイドラインに加えて,家族性腫瘍研究会の「家族性腫瘍における遺伝子診断の研究とこれを応用した診療に関する
ガイドライン」に準拠する.
(3)家族性腫瘍の易罹患性検査を行うにあたっては,検査の感度,特異度,陽性・陰性結果の正診率などが十分なレベルにあることを確認する.
④薬物に対する反応性の個体差を判定することを目的とする遺伝学的検査
薬物代謝酵素の遺伝子多型検査による薬剤感受性診断は,直接治療に役立て得る情報であり,有用性が高いと考えられるが,この情報が社会的差別などに誤用されることのないよう,他の目的の遺伝学的検査と同様の注意が必要である.
⑤出生前検査と出生前診断
(1)妊娠前半期に行なわれる出生前検査および診断には,羊水,絨毛,その他の胎児試料などを用いた細胞遺伝学的,遺伝生化学的,分子遺伝学的,細胞・病理
学的方法,および超音波検査などを用いた物理学的方法などがある.
(2)出生前検査および診断として遺伝学的検査および診断を行うにあたっては,倫理的および社会的問題を包含していることに留意し,特に以下の点に注意して実
施する.
◦胎児が罹患児である可能性(リスク),検査法の診断限界,母体・胎児に対する危険性,副作用などについて検査前によく説明し,十分な遺伝カウンセリングを
行うこと.
◦検査の実施は,十分な基礎的研修を行い,安全かつ確実な検査技術を習得した産科医により,またはその指導の下に行われること.
(3)絨毛採取,羊水穿刺など,侵襲的な出生前検査・診断は,下記のような場合の妊娠について夫婦からの希望があり,検査の意義について十分な理解が得られ
た場合に行う.
◦夫婦のいずれかが,染色体異常の保因者である場合 .
◦染色体異常症に罹患した児を妊娠,分娩した既往を有する場合.
◦高齢妊娠の場合 .
◦妊婦が新生児期もしくは小児期に発症する重篤なX連鎖遺伝病のヘテロ接合体の場合 .
◦夫婦のいずれもが,新生児期もしくは小児期に発症する重篤な常染色体劣性遺伝病のヘテロ接合体の場合 .
◦夫婦のいずれかが,新生児期もしくは小児期に発症する重篤な常染色体優性遺伝病のヘテロ接合体の場合 .
◦その他,胎児が重篤な疾患に罹患する可能性のある場合.
(4)重篤なX連鎖遺伝病のために検査が行われる場合を除き,胎児の性別を告げない.
(5)出生前診断技術の精度管理については,常にその向上に努める.
(6)母体血清マーカー検査の取り扱いに関しては,厚生科学審議会先端医療技術評価部会出生前診断に関する専門委員会による「母体血清マーカー検査に関
する見解」,日本人類遺伝学会倫理審議委員会による「母体血清マーカー検査に関する見解」,および日本産科婦人科学会周産期委員会による報告「母体血
清マーカー検査に関する見解について」を十分に尊重して施行する.
(7)着床前検査および診断は極めて高度な知識・技術を要する,いまだ研究段階にある遺伝学的検査を用いた医療技術であり,倫理的側面からもより慎重に取り
扱う.実施に際しては,日本産科婦人科学会会告「着床前診断に関する見解」に準拠する.
③ 先天性心血管疾患
1)遺伝的要因
旧厚生省班研究の全国調査(1986年)71)では,日本における先天性心血管疾患の発生頻度は生産児の1.06%であり,新生児死亡の原因となる先天性疾患の中では最も頻度が高い.一方,日本小児循環器学会疫学委員会の調査(2003年)72)では,その成因による内訳は,染色体異常(Down症候群,Turner 症候群,22q11.2欠失症候群,Williams症候群など,8.2%)や遺伝子病(Noonan症候群,Holt-Oram症候群,Marfan症候群,遺伝性QT延長症候群など,4.7%)などの遺伝要因によるものが全体の約12.9%,母体の全身疾患,胎内感染,催奇形因子など,環境(外的)要因によるものが0.5%であり,残りの86.7%は成因不明の多因子遺伝(多くの先天性心疾患,特発性肺高血圧症,特発性心筋症など)によると報告されている.多因子遺伝とは,個人の遺伝的素因(遺伝子異常)と環境(外的)因子との相互関係によって疾患が発生するものを指す.すなわち,純粋にメンデル遺伝のみで説明できる先天性心血管疾患は一部であり,大半は胎内での形態形成,生後の発達・発育,加齢に伴う環境要因によって疾患遺伝子の関与の程度が左右され,表現形の多様性と深くかかわっていることが明らかになってきた73)-76).一方,近年のゲノム医学ならび遺伝子改変マウスを用いた発生工学の目覚ましい発展により,多くの遺伝性先天性心血管疾患の責任遺伝子が明らかになってきた.その代表的な疾患と表現型および疾患遺伝子を表5に掲載する73)-75),77),78).各疾患の臨床像や妊娠・出産における問題点に関しては,本ガイドライン後半の各論の項を参照されたい.また,先天性心血管疾患における遺伝的要因に関する詳細は,American Heart Association Congenital Cardiac Defects Committeeより出された声明78),およびボストン小児病院より出されているホームページCongenital Cardiovascular Genetics Program 77)を参照されたい.
2)環境要因
また先天性心血管疾患の成因には,遺伝子異常のみならず,妊娠中の胎児および母体の環境要因も重要である79).妊婦の喫煙は児の心室中隔欠損症,Fallot四徴症,心房中隔欠損症,完全大血管転位症の発症の危険因子であることが,また妊婦の飲酒は中枢神経系の障害を伴う胎児性アルコール症候群とともに,Fallot四徴症,心房中隔欠損症,完全大血管転位症の発症と関連することが報告されている72),80).また,母体が妊娠早期に風疹ウイルスに感染すると,先天性風疹症候群として,先天性白内障や先天性難聴とともに心室中隔欠損症,心房中隔欠損症,肺動脈狭窄症が高率に発症する79).血糖値のコントロールが不良な糖尿病の母体からは,肥大型心筋症,内臓錯位症候群,房室中隔欠損症,完全大血管転位症などが発症する79).またSLEやシェーグレン症候群に罹患した妊婦では,抗SSA,抗SSB抗体が経胎盤的に移行して児に高度房室ブロックが発症することがあり,治療として1度房室ブロック段階での母体へのステロイド投与が推奨されている81),82).フェニルケトン尿症では,高フェニルアラニン血症によりFallot四徴症,心室中隔欠損症,心房中隔欠損症,大動脈縮窄症,左心低形成症候群などが発症する.妊娠前からの低フェニルアラニン食によるコントロールが必要となる83).また,胎児に先天性心血管疾患をもたらす可能性のある薬剤として,アンフェタミン(心室中隔欠損症,動脈管開存症,心房中隔欠損症など),ヒダントイン(肺動脈狭窄症,大動脈狭窄症,大動脈縮窄症など),トリメタジオン(完全大血管転位症,Fallot四徴症,左心低形成症候群など),リチウム(Ebstein病,三尖弁閉鎖症など)が報告されている73)-75),79).妊婦が摂取するビタミンの過不足も先天性心血管異常と深い関係がある.ビタミンAの過剰摂取は胎児に,心臓流出路障害とともに中枢神経系や口蓋や胸腺の異常を来たす76),84).妊婦の葉酸摂取が不足すると胎児に心室中隔欠損症などの心血管異常を来たすため,食品への葉酸の添加もなされている76),79),85),86).母体の有機溶媒への曝露も胎児に房室中隔欠損症,完全大血管転位症,肺動脈狭窄症など様々な先天性心疾患をもたらす79).
3)各疾患群にみられる遺伝子異常
①先天性心疾患
先天性心疾患の多くは多因子遺伝と考えられており,メンデル遺伝様式には従わないことの方が多い.しかしながら,先天性心疾患を持つ親から生まれる子供たちは先天性心疾患を繰り返す頻度が高く,一般頻度に比べて3〜5倍と考えられている72),74),76).この場合,母親が先天性心疾患の場合,父親が先天性心疾患である場合より頻度は2倍以上高い.日本小児循環器学会の全国調査によると,親子間での疾患一致率は,心房中隔欠損症(66.7%)および動脈管開存症(66.7%)が最も高く,次いで心室中隔欠損症(47.1%),Fallot四徴症(28.6%)である72).一方,最近の発生生物学ならび遺伝子工学の目覚ましい発展により,先天性心疾患の原因となる数多くの遺伝子異常が明らかになってきた.前述の表5に原因が明らかになった代表的な遺伝性先天性心血管疾患を示した7),9).
②特発性心筋症
肥大型心筋症の約50%に常染色体性優性遺伝による家族歴が認められる87).ミオシン,アクチン,トロポニン,トロポミオシンなどのサルコメア収縮に直接関連す
る要素以外にも,タイチンなどの細胞骨格や膜蛋白カベオリンなどのシグナル伝達物質の異常でも肥大型心筋症が発症することが明らかとなり87),88)(表6),筋細胞肥大の原因として筋収縮におけるカルシウム感受性の亢進が考えられている89).家族性肥大型心筋症では,約50%に遺伝子異常が明らかにされている.その中では,心筋βミオシン重鎖(MHY7)での変異が約20%,心筋トロポニンT(TNNT2)および心筋ミオシン結合蛋白(MYBPC3)での変異が各々約10%であり,これらの3
種類の家族性肥大型心筋症の主要な原因遺伝子となっている.その中では,MYH7 およびMYBPC3の変異例では肥大の程度が強く,TNNT2 の変異例では肥大の程度は軽いが心収縮力の低下し比較的早期より拡張相に移行する.弧発例の肥大型心筋症おいても約15%に変異が見つかる87),88)(レベルC).
肥大型心筋症類似の病態を示す疾患として,多くの蓄積病の原因遺伝子も明らかになってきた.Pompe病(α1,4- グルコシダーゼ欠損),Danon病(リソゾーム膜蛋
白欠損症),Fabry病(αガラクトシダーゼA欠損症),AMP活性化蛋白キナーゼ変異症などでは心肥大がみられる87),88),90).
拡張型心筋症の家族内発症は肥大型心筋症に比べて少なく,約30%とされている.原因遺伝子としては,αアクチニン,デスミン,タイチン,ジストロフィンなど,心筋細胞骨格や細胞接着分子に変異が認められることが多いが,肥大型心筋症同様にサルコメア変異も報告されている87),88),91) (表6).不整脈原性右室心筋症は,臨床的に右心室に期限を有する心室性不整脈を主症状とし,病理組織学的に右心室優位の心筋細胞の変性,脱落と線維脂肪浸潤の増加を示す疾患である.遺伝子異常として,約30%の症例にプラコフィリン2(PKP2)が,またその他に,デスモプラキン,プラコグロビン,プラコフィリン,リアノジン受容体-2,TGFβ 3などの遺伝子変異が報告されている92),93)(レベルC).
ミトコンドリア心筋症は,ミトコンドリアにおけるエネルギー産生障害に基づいて引き起こされるミトコンドリア病の中で,肥大型心筋症,拡張型心筋症,拘束型心筋症,刺激伝導障害などの多様な心病変を示す代謝性心筋症である95),96).一般にミトコンドリア病では,中枢神経障害(精神運動発達遅滞,痙攣,意識消失),筋力低下,乳酸アシドーシスなどの全身症状がみられることがある.ミトコンドリア心筋症は母系遺伝を示すことが多く,無症状の母方家族に変異が検出されることがあり,遺伝学的な配慮が必要である.
これらすべての遺伝性心筋疾患においては,妊娠に伴う循環動態の変化により生じる母体および児へのリスクを十分に説明するとともに,児への疾患の繰り返しに関する遺伝的情報を説明する必要がある.
③遺伝性不整脈
突然死の原因となる遺伝性QT延長症候群,カテコラミン誘発多形性心室頻拍,Brugada症候群,進行性心臓伝導障害,家族性洞機能不全症候群,家族性心房細動,QT短縮症候群などにおいて,遺伝子変異が明らかとなっている96)-98)(表7).
QT延長症候群は,基礎疾患がみられずに心電図上QT間隔が延長し,心室頻拍や心室細動から失神および突然死に至る可能性のある疾患群である.現在までに,カリウムおよびナトリウムチャネル遺伝子を中心として,LQT1-LQT12まで分類されており,300以上の遺伝子異常が報告されている97)-99).その内訳は,LQT1(約40%),LQT2(約30〜40%),LQT3(約10%)とされており,LQT1〜LQT3で全体の約90%を占める.一般にLQT1は運動中や水泳中に,LQT2では睡眠中の聴覚刺激(目覚まし時計など)により,LQT3 では安静時および睡眠中に心事故を起こす.
Brugada症候群は,右側胸部誘導心電図(V1-V3)にて右脚ブロック様の心電図波形と特徴的なST上昇(coved 型またはsaddle back型)を伴う,中年男性に多い特発性心室細動の症候群である.責任遺伝子としてナトリウムチャネルαサブユニット(SCN5A)の異常が15〜25%に報告されている100).
カテコラミン誘発多形性心室頻拍は,学童や青年期において運動や交感神経の刺激に伴い心室頻拍から失神や突然死を来たす症候群であり,心筋細胞小胞体のリアノジン受容体(RyR2)の遺伝子異常が報告されている101).
遺伝性不整脈疾患の内服治療の詳細および植込み型除細動器の適応については,日本循環器学会「QT延長症候群(先天性・二次性)とBrugada症候群の診療に関するガイドライン(2007年)」102)を参照されたい.
これらすべての遺伝性不整脈疾患においても,妊娠に伴う母体および児へのリスクを十分に説明するとともに,児への疾患の繰り返しに関する遺伝的情報をカウン
セリングする必要がある.
④その他の先天性心血管疾患
22q11.2欠失症候群,Marfan症候群,Loeys-Dietz症候群,Ehlers-Danlos 症候群,原発性肺高血圧症,Eisenmenger症候群,先天性心疾患術後遺残病変,Fontan手術後などにおいては,妊娠・出産に際して生じる母体の循環動態の変化に伴い,母親と胎児の厳重な管理が必要である.また,児への疾患の繰り返しに関する遺伝的情報を説明する必要がある.
表5-1 代表的な遺伝性および染色体異常による先天性心血管疾患
表5-2 代表的な遺伝性および染色体異常による先天性心血管疾患
表6 遺伝性心筋症の疾患遺伝子
AD:常染色体優性遺伝,AR:常染色体劣性遺伝,XR:X染色体劣性遺伝
表7 不整脈疾患にみられる遺伝子異常
- Home
- Ⅱ 総論
- 3 妊娠カウンセリング
- 5 遺伝カウンセリング
心疾患患者の妊娠・出産の適応、管理に関するガイドライン(2010年改訂版)
Guidelines for Indication and Management of Pregnancy and Delivery in Women with Heart Disease (JCS 2010)
Guidelines for Indication and Management of Pregnancy and Delivery in Women with Heart Disease (JCS 2010)